“Children today born with hemophilia are being born into an era where, due to these groundbreaking therapies, they should in all respects be able to live their lives to their fullest potential.”
— Steven Pipe, MD, Chair, NHF Medical and Scientific Advisory Council
IN OUR LIFETIMES, no serious medical condition has had its prognosis more dramatically transformed by scientific advances than severe hemophilia. Prior to the first use of factor-enriched cryoprecipitate and plasma-based factor VIII (FVIII) and factor IX (FIX) concentrates in the 1960s, followed by recombinant factor products in the 1990s, clinicians were limited to transfusions of fresh frozen plasma to try to limit bleeds into joints, soft tissues and the central nervous system. Children with severe disease (defined as a FVIII or FIX level of less than 1 percent of normal) grew up experiencing frequent painful joint bleeds resulting in crippling joint hemarthroses. The ever-present risk of catastrophic spontaneous or trauma-induced hemorrhage translated into much-shortened lives. As recently as 1960, the average lifespan of a male with severe hemophilia was less than 20 years.1
By the mid-1990s, “on-demand” factor replacement therapy to manage frequent bleeds began to give way to a far more effective strategy: prophylactic dosing, typically every other day or three times weekly, of a sufficient dose of replacement factor to maintain circulating levels high enough to protect against spontaneous and excessive trauma-induced bleeding. Approved starting in 2014, the newest generation of FVIII and IX concentrates are structurally or chemically altered to extend their circulating half-life, significantly reducing the frequency of infusions to maintain protective levels. Today, a child with moderately severe or severe hemophilia A or B who is fully adherent to prophylactic treatment regimen has a much-reduced risk of serious bleeding episodes, with the potential to attain a normal life expectancy.2
The disease burden of severe hemophilia in the U.S. and other developed countries is now largely associated with 1) the demands of complying with regular prophylactic infusions of factor concentrate over a lifetime, 2) limitations on physical activities that can still induce joint, soft tissue or central nervous system bleeds and 3) a roughly 30 percent risk, usually in early childhood, of developing inhibitor alloantibodies that neutralize exogenously administrated clotting factor, leaving the patient exposed to potentially life-threatening bleeds. A significant residual bleeding risk remains, however, as mean coagulation factor levels attained with prophylaxis are still far below the normal range and reach a nadir in the latter period prior to the next infusion. Finally, there are exceedingly high annual costs of prophylactic therapy with factor replacement therapies, which can amount to tens of millions of dollars over a lifetime. All of these considerations have encouraged the development of potentially curative gene therapy.
How Hemophilia Gene Therapy Works
The principle of gene therapy is straightforward: resolve or at least ameliorate disease caused by a defective gene by populating target cells with the normal gene that encodes and expresses therapeutic levels of functional protein. But there are two fundamentally different ways to do it. GlaxoSmithKline’s Strimvelis gene therapy, approved in the European Union in 2016 for a variant of severe combined immunodeficiency, is a pioneering example of cell-based delivery, wherein the patient’s bone marrow or circulating CD34+stem cells are harvested and cultured with a viral vector that inserts the corrective gene directly into the stem cell cDNA; these expanded gene-corrected stem cells are then reintroduced into the patient. Thus, the gene therapy “product” is the patient’s own genetically engineered autologous cells.
But ex vivo genetic manipulation of harvested stem cells is complex and requires the cells be transported to a centralized laboratory with this expertise for processing. In addition to Strimvelis, investigational gene therapy programs addressing hematological disorders such as sickle cell disease and β-thalassemia are other examples of cell-based delivery of gene therapy involving stem cell harvesting and ex vivo manipulation.
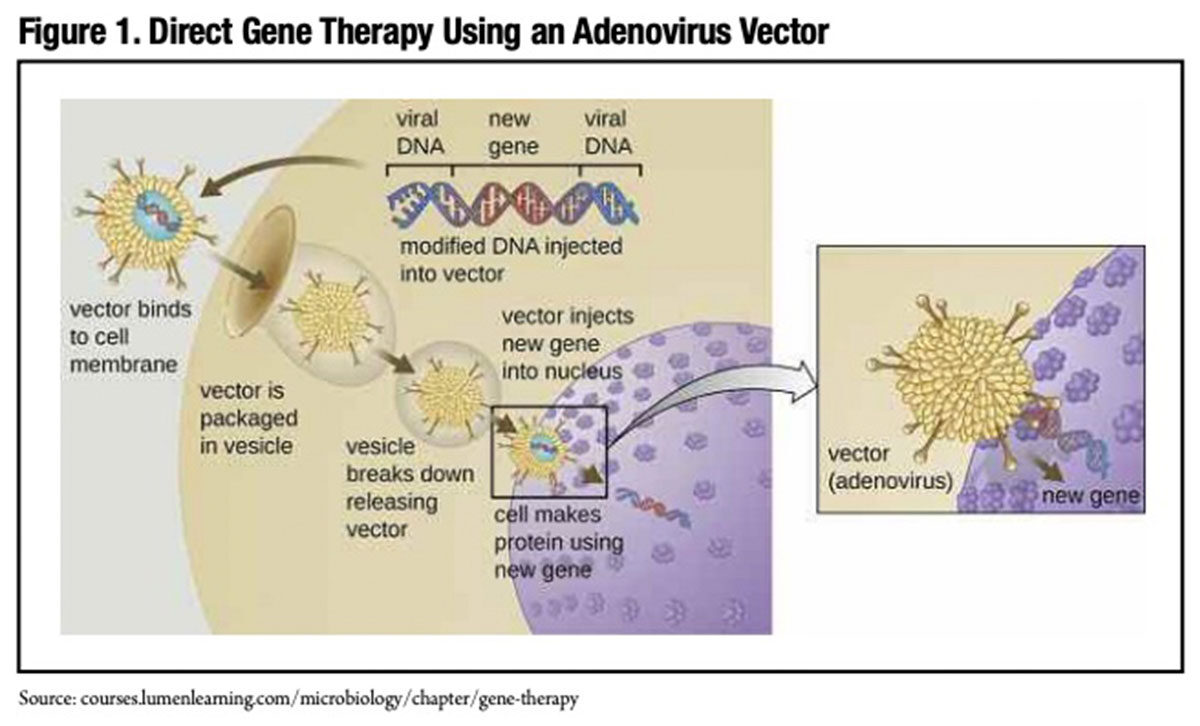
In contrast, current development-stage hemophilia gene therapies deliver transgenes encoding FVIII or IX directly to the patient’s target liver cells (Figure 1). Large numbers of these transgenes, which are ferried into target liver cells by an adenoassociated virus (AAV) vector containing a “liver-specific promoter,” are intravenously infused in a single treatment session. The first approved gene therapy product in the U.S., Spark Therapeutics’ LUXTURNA,* is a directly delivered gene therapy. Not unlike any other approved biotherapeutic, LUXTURNA is manufactured at a U.S. Food and Drug Administration (FDA)- licensed facility in Philadelphia. It is purchased by the hospital and supplied to the operating room, where it is injected subretinally. Similarly for a future hemophilia gene therapy, a manufactured AAV-FVIII or IX transgene would simply be supplied to the hospital and administered to the patient.
Hemophilia gene therapy differs in one other important respect from ex vivoprocessed stem cell gene therapies. The AAV vector delivers the FVIII or IX gene to the liver cell nucleus, but in this instance, it typically does not integrate into the cellular DNA. Instead, the transgenecontaining vector remains separate from the chromosomes. Because the AAV does not integrate itself into the cellular DNA with investigational hemophilia gene therapies, the risk of potentially creating cancer-inducing mutations (known as insertional mutagenesis) that has occurred with investigational ex vivo stem cell-based gene therapies is minimized.
A Head Start for Hemophilia B Gene Therapy
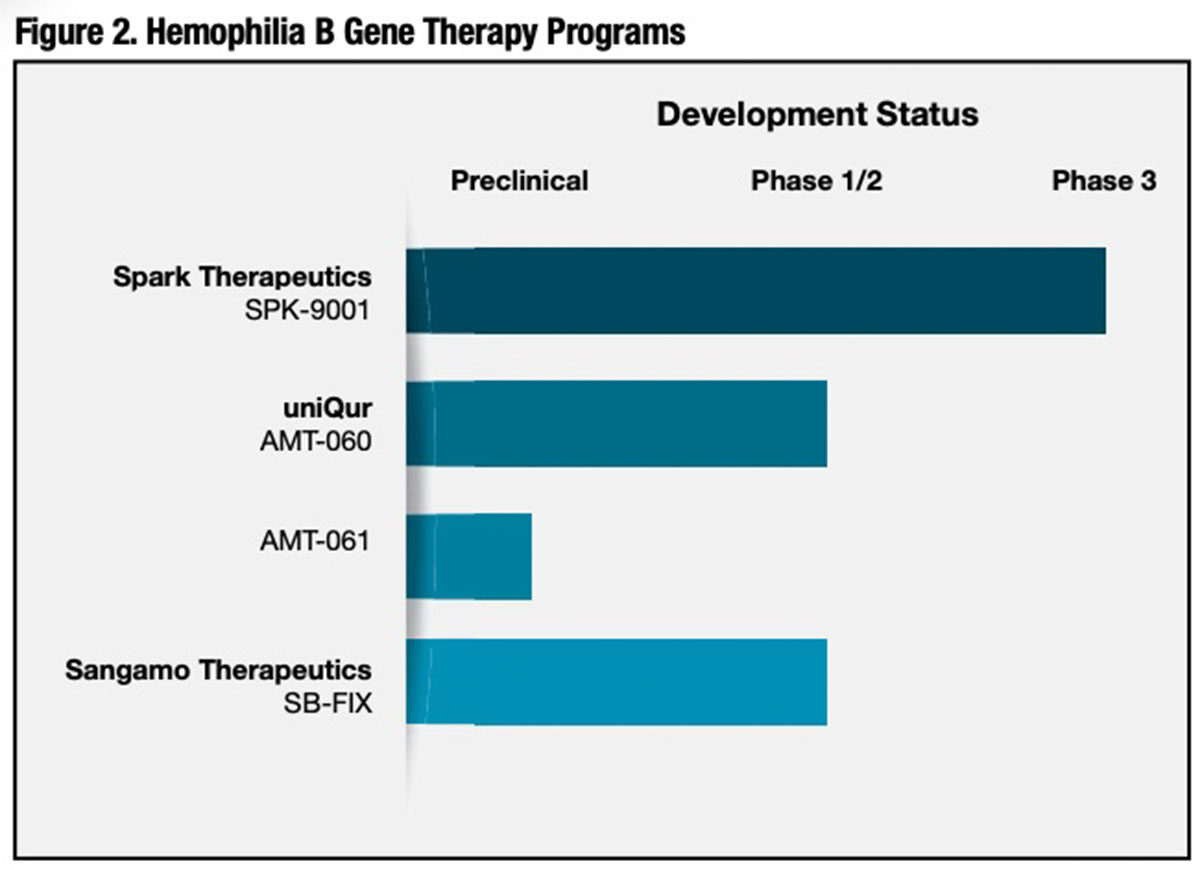
While there are six times as many individuals with severe hemophilia A than with severe hemophilia B, early gene therapy efforts focused on hemophilia B for the simple reason that the gene DNA encoding FIX is about half as large as the DNA encoding FVIII. Until very recently, the comparatively large size of FVIII DNA was widely thought to be problematic given the limited packaging capacity of AAV capsids. 3 The gene coding FIX is in fact easier to incorporate into various viral vectors with robust protein expression in vivo. By 2013, after advancing through preclinical testing, three investigational hemophilia B gene therapy products had entered early-stage clinical trials in patients with severe or moderate-severe hemophilia B (Figure 2). While numbers of treated subjects are small, recently presented findings from these studies are very encouraging.
Spark Therapeutics/Pfizer (SPK-9001). This novel gene therapy product incorporates a FIX Padua transgene in its recombinant AAV vector. U.S., Canadian and Australian investigators infused SPK-9001 at a dose of 5 x 1011 vector genomes per kilogram (vg/kg) of body weight in 10 adult male subjects previously on prophylaxis, all of whom had baseline FIX coagulant activity of ≤2 percent of the normal value. On follow-up ranging from 28 weeks to 78 weeks, the annualized bleeding rate (ABR) was reduced from a mean of 11.1 events per year (range 0 to 48) to a mean of 0.4 events per year (range 0 to 4) after vector administration. All 10 subjects experienced sustained elevations in their FIX coagulant activity, with a mean steady-state of 33.7 ± 18.5 percent of normal (range 14 percent to 81 percent).
FIX concentrate use in these subjects correspondingly fell from a mean monthly dose of 2,908 IU per kg (range 0 IU to 8,098 IU) prior to SPK-9001 administration to just 49 IU per kg (range 0 IU to 376 IU) after administration. Eight of the 10 subjects didn’t require any exogenous FIX, and nine of 10 experienced no bleeding events after vector administration. No clinically significant adverse events were observed.
In their report, investigators concluded that a single administration of SPK-9001 gene therapy “enabled the termination of baseline prophylaxis and the near elimination of bleeding and factor use.”4
uniQure (AMT-060). A single administration of the higher of two evaluated doses (2 x 1013 genome copies) of uniQure’s investigational AAV serotype 5 (AAV5)-human FIX hemophilia B gene therapy reduced the mean annualized spontaneous bleeding rate from 3.0 to 0.9 bleeds in five adult subjects with endogenous FIX levels ≤2 percent of normal and a severe bleeding phenotype. 5 Annualized FIX use decreased by 73 percent, and eight of nine participants receiving FIX at study entry were able to stop prophylaxis. FIX expression levels were stable over one-and-a-half years of follow-up, and there were no serious adverse events or development of inhibitors. However, at the highest tested dose, AMT-060 does not appear to effect as robust an elevation in endogenous FIX production as competitive hemophilia B gene therapies currently in clinical development.
In late 2017, this Netherlands-based biotechnology firm reported that AMT061, a modified version of its original hemophilia B gene therapy, achieved an approximately 6.5-fold improvement in FIX activity in cynomolgus monkeys. 6 Like Spark Therapeutics’ gene therapy product for hemophilia B, AMT-061 substitutes the high-activity FIX-Padua gene variant for wild-type FIX in the AAV5 capsid-based vector. While expression of FIX is similar to AMT-060, animals infused with a single dose of AMT-061 had approximately 6.5-fold higher FIX activity. Moreover, at a dose of 5 x 1012 vg/kg, FIX activity averaged 58.9 percent of normal from week 4 to week 13 postadministration, compared to an average of just 9.1 percent of normal for AMT-060. uniQure plans to advance AMT-061 into a pivotal clinical study in 2018.
Sangamo Therapeutics (SB-FIX). Sangamo has taken a unique “gene editing” approach to gene therapy for severe hemophilia B using a site-specific zinc finger nuclease (ZFN) to integrate the FIX transgene within the albumin gene of the liver cell genome. 7 An AAV vector is additionally used in vivo to deliver the FIX gene. Sangamo targeted the albumin gene because of liver cells’ very large capacity to produce this blood protein — about 15 grams per day. The company believes that targeting and co-opting only a very small percentage of the albumin gene’s capacity could potentially produce therapeutically relevant levels of FIX with no significant effect on albumin production. 8
Sangamo’s Phase I/II “FIXtendz” trial currently in progress will enroll 12 subjects with severe hemophilia B to evaluate the safety, tolerability and preliminary efficacy of three escalating doses of SB-FIX.
Hemophilia A Gene Therapy Programs Advance
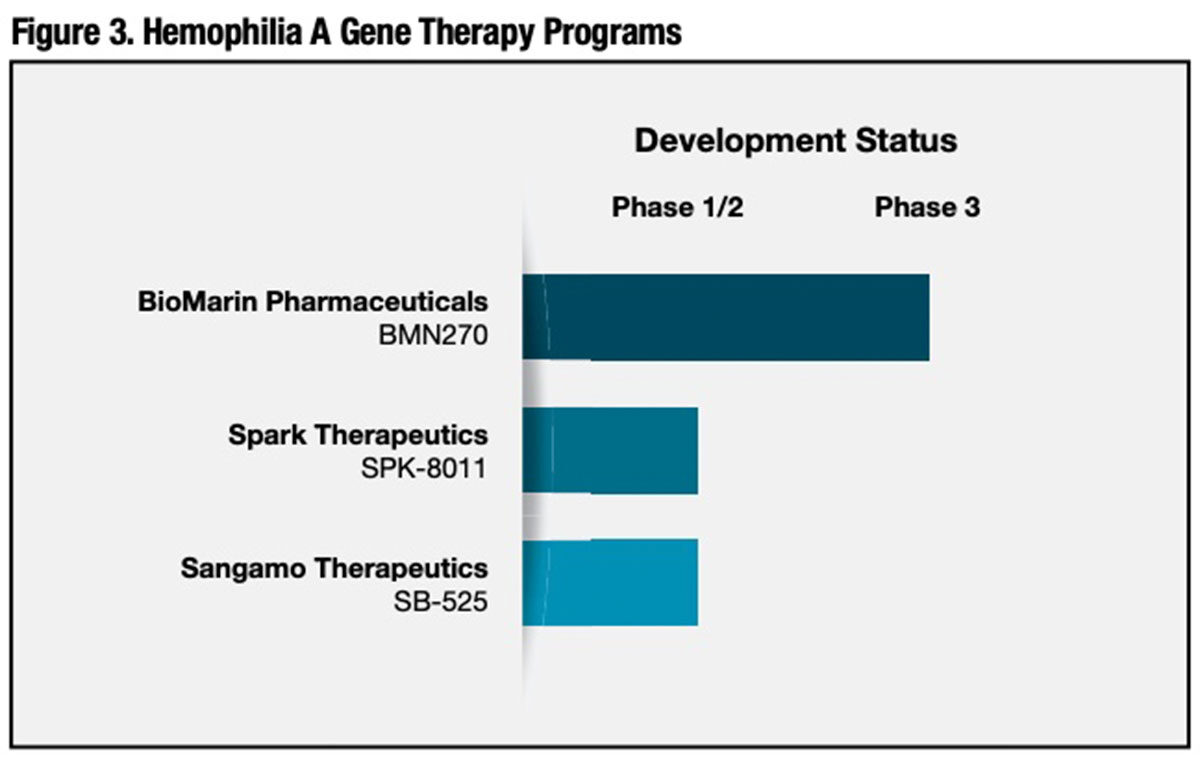
Despite the head start for investigational hemophilia B gene therapies to enter clinical trials, one U.S. competitor, BioMarin Pharmaceuticals, has managed to close the gap with its proprietary hemophilia A gene therapy, and is now enrolling the first subjects in a pivotal Phase III trial. Two others, Spark Therapeutics and Sangamo Therapeutics, are currently enrolling patients in Phase I/II studies (Figure 3).
BioMarin Pharmaceuticals (BMN 270). Founded 20 years ago, BioMarin has already secured FDA approval for several enzymes and drug compounds to treat rare congenital disorders, and has several others in the development pipeline. BMN 270 or valoctocogene roxaparvovec, intended for treatment of severe hemophilia A, is the company’s sole gene therapy candidate. BMN 270 comprises an AAV5 vector encoding a B domain-deleted FVIII gene.
In initial mouse models of hemophilia A, BMN 270 restored FVIII plasma concentrations to levels projected to be adequate for normal clotting activity in humans. In Phase I/II clinical trial findings reported last December, BioMarin reported the sustained normalization of FVIII activity level over a period of one year in six of the seven study participants receiving the highest of three doses of BMN 270 (6 x 1013 vg/kg). All seven highdose subjects experienced stabilization of hemostasis and a profound reduction in FVIII use. 9
At 78 weeks following BMN 270 treatment, the mean and median circulating FVIII levels for the high-dose patient cohort were, respectively, 90 percent and 89 percent of normal. In all seven subjects, the median ABR dropped from 16.5 bleeds prior to BMN 270 therapy to zero bleeds (mean 16.3 to 0.5 bleeds). Annualized FVIII infusions per subject dropped from a median of 138.5 infusions prior to gene therapy to zero infusions (mean 136.7 to 6.1 infusions). Remarkably, none of the seven subjects in the high-dose cohort used any FVIII for self-reported bleeds between week 22 and the end of the study period. Prior to gene therapy, the median FVIII utilization in this cohort was about 400,000 IU per year.
As of mid-November 2017, no subject receiving BMN 270 developed inhibitors to FVIII. Adverse events were considered minor; eight of the nine study subjects experienced mild elevations in serum alanine aminotransferase that resolved without sequelae. While these results are obviously highly promising, the investigators caution that continued follow-up is needed to determine the long-term safety and efficacy of BMN 270.
In December 2017, BioMarin enrolled the first of a planned 40 patients with severe hemophilia A in its global “GENEr8-1” Phase III clinical trial to evaluate the efficacy and safety of a single 6 x 1013 vg/kg dose of BMN 270. A second Phase III trial scheduled to start in early 2018, “GENEr8-2,” will separately evaluate a single 4 x 1013 vg/kg dose of BMN 270.
Spark Therapeutics (SPK-8011). As of last December, early outcomes had been reported on seven severe hemophilia A patients dosed with a single infusion of 5 x 1011 vg/kg of this liver-targeted, human FVIII gene-containing AAV-LK03 capsid. After an initial four-week induction period, the overall annualized infusion rate of exogenous FVIII was reduced by approximately 98 percent, to a mean of just 1.2 infusions from a baseline mean of 57.8 infusions prior to gene therapy.
In two participants followed for more than 30 weeks, one has achieved sustained FVIII expression with a mean activity level of 10 percent of normal after week 12, while the other has demonstrated much more variable expression kinetics, with a mean FVIII level of 16 percent of normal, but with fluctuation ranging from 6 percent to 37 percent. Preliminary findings of this nature clearly underscore the importance of results from long-term follow-up of larger numbers of treated subjects for this and all other investigational gene therapies.
Sangamo Therapeutics (SB-525). Sangamo recently initiated a Phase I/II clinical trial of SB-525, a recombinant AAV2/6 vector encoding the gene for B-domain deleted human FVIII. The expressed protein has the same amino acid sequence as Pfizer’s XYNTHA, a recombinant FVIII product approved for the treatment of hemophilia A.
Potentially Favorable Cost Economics
Appropriately, enthusiasm for hemophilia gene therapy relates to its potential to translate into better protection against bleeding risks and an improved quality of life. But its eventual approval will bring an additional collateral benefit into focus: cost savings.
“For a 55 kilogram person with severe hemophilia A on prophylaxis, the average annual usage is around 330,000 IUs of standard FVIII per year, assuming he is 100 percent treatment-compliant,” said Matthew Hotchko, PhD, vice president of research at the Connecticutbased Marketing Research Bureau. “At the current price, this translates into a cost of close to $300,000 per year. And, of course, the cost of prophylaxis increases in direct proportion to increased body weight.” The annual average cost for extended half-life products is significantly higher due to the higher price of this new product class, Dr. Hotchko added — about $400,000 to $450,000 for a 55 kg individual with severe disease, again assuming he is 100 percent treatment-compliant.
Approved gene therapies will unquestionably be costly. But in the case of hemophilia, the presumption that these highly sophisticated treatments will boost healthcare spending on the disease is turned on its head. Because gene therapies promise to all but eliminate the need for extraordinarily costly chronic factor replacement therapy, these one-time treatments can be expected to importantly reduce the lifetime cost burden of severe hemophilia A and B.
Who Will Get Hemophilia Gene Therapy?
The task of defining appropriate candidates for a given hemophilia A or B gene therapy will obviously need to await the enrollment and long-term follow-up of a sufficient number of study subjects to provide acceptable clarity about its safety and efficacy. In particular, referral for a given gene therapy product will heavily depend on its success rate and the persistence of protective levels of clotting factor. These answers remain a number of years away.
But let’s assume for a moment that one or more investigational gene therapies prove to be safe and reliably maintain factor activity well above the target levels that currently guide FVIII and FIX prophylaxis schedules. While prophylaxis has dramatically reduced the risk of spontaneous and traumatic bleeds, it is still far from an ideal therapy.
The shortcomings of prophylaxis with factor concentrates extend beyond the known risk of developing inhibitors and the adverse quality-of-life impact of having to regularly self-infuse the product for moderately severe or severe disease.
People on prophylaxis still cannot engage in so-called “high-risk” sports or other physical activities that could potentially induce serious bleeds into the joints, soft tissues or brain. As each scheduled factor infusion approaches, and the factor level drops to its nadir, therisk of an activity- or injury-related bleed, in particular, increases. And, of course, noncompliance or even occasional forgetfulness boosts the chances for a serious or life-threatening bleeding event.
It is widely acknowledged that while chronic prophylactic treatment with factor products has improved patient quality of life and protection against bleeds, it leaves much to be desired. If preliminary evidence is any indication, gene therapy could indeed prove to be the definitive cure that, at last, frees thousands of people with hemophilia to live a normal life.