 If you are of a certain age, you may remember heart-wrenching images and a long-running — and ultimately tragic — story about a Texas boy whose 12 years of life inside a plastic isolation bubble introduced Americans to the plight of children born with severe combined immunodeficiency (SCID). David Vetter, dubbed the “bubble boy” in countless news features, lived in a succession of sterile chambers to avoid the fate of his older brother, who had succumbed in infancy from complications of overwhelming infection from the same disorder.
If you are of a certain age, you may remember heart-wrenching images and a long-running — and ultimately tragic — story about a Texas boy whose 12 years of life inside a plastic isolation bubble introduced Americans to the plight of children born with severe combined immunodeficiency (SCID). David Vetter, dubbed the “bubble boy” in countless news features, lived in a succession of sterile chambers to avoid the fate of his older brother, who had succumbed in infancy from complications of overwhelming infection from the same disorder.
From his mother, David inherited a defective version of the IL-2 common chain receptor gene (IL2RG) that she carried on one of her two X chromosomes. David’s X-linked disorder, SCID-X1, affects only boys and accounts for 40 percent to 50 percent of all SCID cases. Any of at least 300 different mutations can disable the IL2RG gene, which encodes a protein critical for regulating growth and maturation of T and B lymphocytes and other immune cells responsible for killing bacteria, viruses, fungi and other invasive pathogens.
The next most common form, ADA-SCID, accounts for 15 percent to 20 percent of cases and equally affects boys and girls. In this instance, the genetic defect results in a non-functional enzyme called adenosine deaminase (ADA), which, like SCID-X1, leads to profoundly low numbers of T lymphocytes (T cells), B lymphocytes (B cells) and natural killer cells.
Altogether, more than 20 recognized forms of “classical SCID” are characterized by a very low T-cell count with near-absent responsiveness to immunogenic stimuli. These patients not only have profound deficits in cellular immunity, but also have very poor antibody response when they come in contact with bacteria, viruses and other pathogens that infants with normal immune function (for their age) can readily fight off. Without immune reconstitution or the kind of extreme measures used to protect David Vetter, nearly all of these children will die from overwhelming infection by the second year of life.
With an incidence estimated at just one in 30,000 to 60,000 live births, children with SCID are referred to major academic medical centers that have well-trained specialists who can manage infections and order prophylactic antibiotic and immune globulin (IG) therapy, and map out a definitive treatment plan. For most patients with classical forms of SCID, definitive treatment is to attempt to reconstitute the immune system by intravenously administering functional hematopoietic stem cells (HSCs) sourced from donor bone marrow, peripheral blood or umbilical cord blood. Ideally, these cells engraft in the bone marrow and restore cellular and antibody-mediated immunity.
The (Improving) Promise of Cure: Donor HSCT
In 1968, pediatricians at the University of Minnesota were the first to infuse HSCs from the bone marrow of a human leukocyte antigen-matched sibling to achieve immunological correction of an infant with X-linked SCID.1 Over the ensuing four and a half decades, specialists have turned to HSC transplants (HSCTs) as a potentially life-saving treatment for SCID with mixed results. Some infants have experienced partial or complete restoration of immune function, while others have suffered engraftment failure or serious complications, including graft versus host disease (GVHD) and toxicity from preparative immunosuppressive “conditioning.”
A number of factors (Table 1) appear to influence the prospects for successful HSC engraftment and survival. Prominent among them are 1) the type of stem cell donor, 2) the “conditioning” regimen prior to transplantation that facilitates donor cell engraftment, 3) recipient age at transplantation and 4) recipient infection status at transplantation.
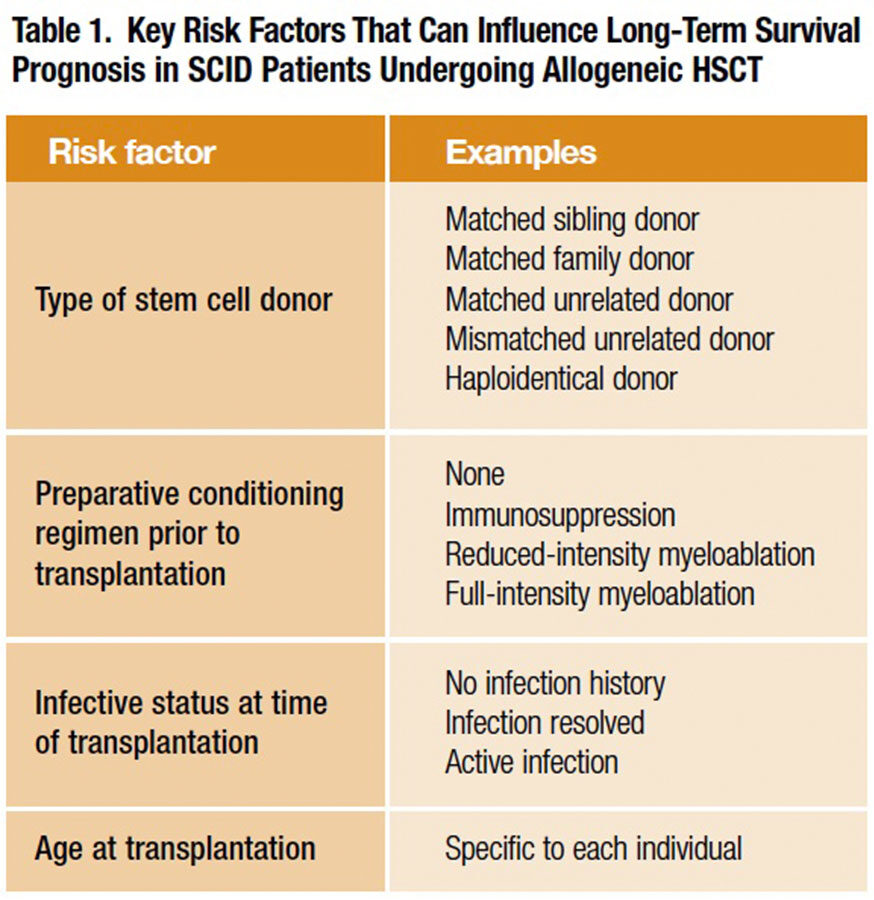
For many years, availability of limited numbers of transplant procedures, together with combinations of these presumptive “risk factors” that vary from one SCID patient to the next, largely frustrated efforts of clinicians to discern which of them importantly impacted long-term survival after HSCT. Finally, a collaborative network of 25 U.S. and Canadian institutions — the Primary Immune Deficiency Treatment Consortium (PIDTC) — tasked itself with retrospectively gathering and analyzing demographic, treatment and long-term survival data from 240 infants with classical SCID who had undergone allogeneic (human donor) HSCT over a 10-year period from Jan. 1, 2000, through Dec. 31, 2009. This large case series, published in 2014, yielded a trove of valuable information to aid HSCT treatment planning.2
Prior to their procedure, 171 of the 240 infants — just more than 70 percent — had already suffered a documented infection. Well over half — 62 percent — of those infected infants were still suffering from their infection at the time of transplantation. Bacterial infections were the most common, followed by the yeast-like fungus Pneumocystis jirovecii (the causative organism for Pneumocystis pneumonia) and respiratory and DNA viruses. Most infants were diagnosed as the result of one or more severe infections from which they struggled to recover, even with the use of aggressive antibiotic therapy.
Most striking about the PIDTC survival analysis (Table 2) was that the entire cohort of infants transplanted under age 3.5 months had the highest long-term survival (94 percent; 64 of 68 children surviving). There was a similar 90 percent five-year survival rate (21 of 23 surviving) for infants older than 3.5 months at transplant who had no history of infection. The five-year survival rate was 82 percent (48 of 58 surviving) in the cohort of infants older than 3.5 months who had experienced a clinical infection that fully resolved by the time of transplantation. But the survival rate plummeted for infants older than 3.5 months who also had an active infection at the time of transplantation: just 50 percent (45 of 91) were alive after five years.
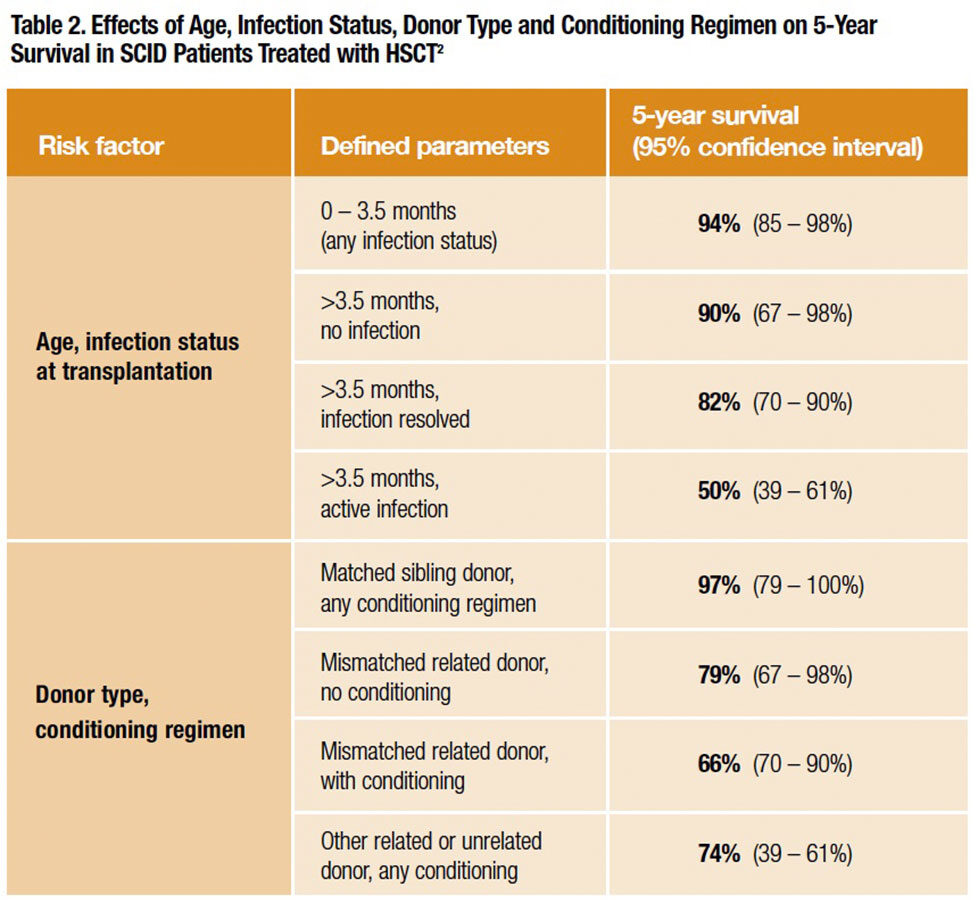
While infants receiving HSCs from a matched sibling donor also had an excellent survival outcome (97 percent), the unfortunate fact is that fewer than 15 percent had access to a matched sibling donor. All the rest of the 240 infants in this case series were forced to accept HSCs from a mismatched related donor (79 percent five-year survival when no immunosuppressive conditioning was used; 66 percent when it was used) or other related or unrelated donors (74 percent five-year survival). By contrast, infants transplanted under age 3.5 months had a very high five-year survival experience despite the fact that the majority received less optimal mismatched related or unrelated donor HSCs, and at least some had a prior infection history or active infection at the time of their procedure.
The PIDTC data convincingly show that infants with SCID should undergo an HSCT procedure within the first 3.5 months of life, ideally before a first severe infectious illness. But as infants with SCID appear entirely healthy at birth, this presents a challenge. Usually nothing appears amiss directly up to the first hospitalization with severe and potentially life-threatening infection, when finally a battery of tests reveals the absence of a functioning immune system. Thus, diagnosis of SCID has historically been reactive — usually after a first or second severe opportunistic infection. The median age of the PIDTC SCID cohort, for example, was nearly 140 days at diagnosis; the median age at transplant was six months, by which time the health of most infants had already been harmed by opportunistic infection.
Newborn TREC Screening Jump-starts SCID Therapy
Seemingly on cue, a practical population-based newborn screening for SCID arrived to detect this occult disorder: the T-cell receptor excision circle (TREC) assay. First used in patients with HIV and hematological malignancy, the TREC assay was adapted to utilize the dried blood spots already universally obtained by heel-stick from infants in the first days of life to screen for metabolic diseases, cystic fibrosis, hypothyroidism and hemoglobin disorders. The TREC copy number is a biomarker for the output of T lymphocytes (lymphopoiesis) from the thymus. A very low TREC value identifies infants with SCID, who have profoundly decreased circulating naïve T cells, SCID-like disorders including “leaky SCID” and Omenn syndrome, and other non-SCID conditions associated with low T-cell counts.
How reliable is the TREC assay for identifying the rare case of SCID among the many thousands of unaffected babies? In 2008, Wisconsin became the first state in the U.S. to screen all newborns. The results of infant screening over the first three years (Figure 1) essentially tell the story. The specificity of the TREC assay — the proportion of healthy individuals correctly identified as test-negative — was a remarkable 99.98 percent. This is important, as too many “false positives” would unnecessarily create parental worry and drive up costs for fruitless additional testing. Equally if not more important was its 100 percent sensitivity: TREC screening identified every case of SCID.3
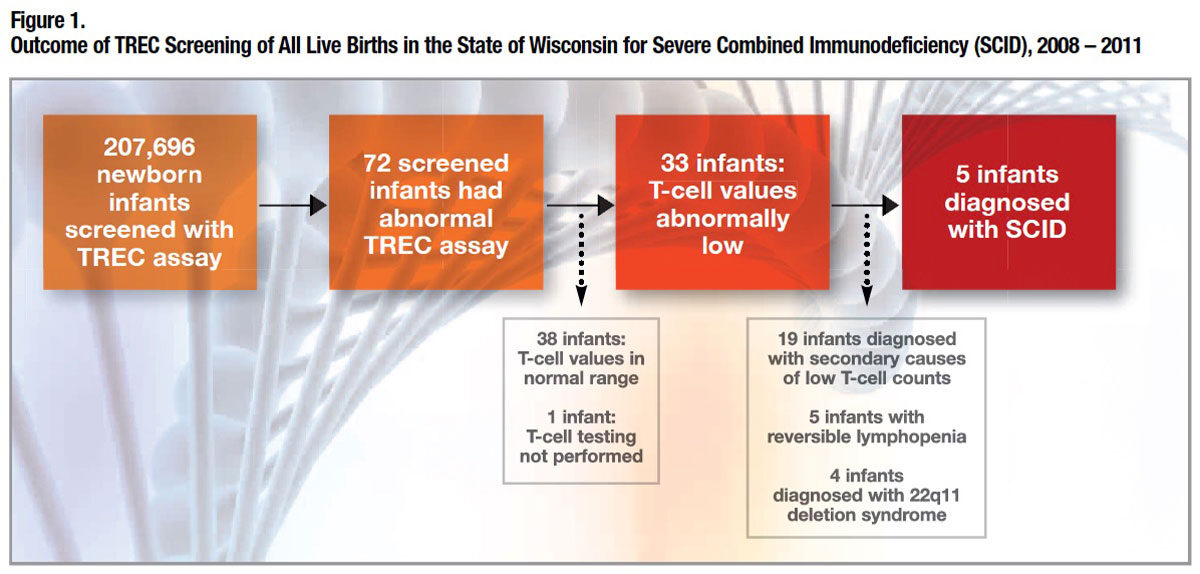
Of the 207,696 infants tested, 0.035 percent had an abnormal TREC screen. On further testing, normal T-cell counts were found in 53 percent of the infants with abnormal screen, but the remainder had varying levels of T-cell lymphopenia (low T-cell count). Of the patients with severe T-cell lymphopenia identified by the TREC assay, further testing determined that 58 percent had various secondary causes (e.g., a congenital anomaly, lymphatic abnormality or metabolic disorder), 15 percent had reversible T-cell lymphopenia, 12 percent had 22q11 chromosomal deletion syndrome and 15 percent — five newborns — had laboratory-confirmed SCID. Four of those five were referred for HSCT, and the fifth (with ADA-SCID) was placed on adenosine deaminase (ADA) replacement therapy.4 At last report, all five SCID patients are alive, and those who received a donor stem cell transplant are alive and well.5
In May 2010, the U.S. Secretary of Health and Human Services added the TREC assay to the Uniform National Newborn Screening Panel. Unrelenting advocacy by the Jeffrey Modell Foundation, the Immune Deficiency Foundation and other groups has convinced 42 states to adopt TREC newborn screening, of which 28 are already up and running and 14 others expect to be sometime in 2015.6 Just eight states — Alabama, Arizona, Indiana, Kansas, Louisiana, Montana, Nevada and Vermont — have yet to approve, fund and implement TREC screening for all newborns.
Transplants for PI Disorders Other Than SCID
The uniform lethality of SCID in infancy demands that clinicians and families accept some risks in pursuit of a realistic potential cure. This same principle applies for a number of other rare primary immunodeficiency (PI) disorders. Prominent among those often treated with HSCT are the following:
• Chronic granulomatous disease (CGD) is an inherited disorder of neutrophil function caused by mutations of an enzyme critical for phagocytic killing of intracellular pathogens. Severe and prolonged infections of the lungs, lymph nodes and skin are frequently found at diagnosis. Life expectancy is short, with only about one-half of affected individuals still alive by age 30.7 First performed in the mid-1980s, HSCT remains the only curative therapeutic option.
While selection of which CGD patients would benefit from HSCT is still debated, those with signs and symptoms suggesting a guarded prognosis may be the most appropriate candidates.8 After HSCT using well-matched donors, 18 of 20 children and young adults in a recent case series were alive at four to 117 months (median 61 months), with normal neutrophil function. Colitis affecting 10 of these patients resolved, and all seven with growth failure experienced catch-up growth.
• Wiscott-Aldrich syndrome (WAS), an X-linked recessive disorder that affects about four in every one million male births, is associated with a life expectancy of less than 20 years. Most patients die of infections, malignancy, autoimmune-related illness or bleeding complications.9
Five-year survival following HSCT, which is the only curative therapy for WAS, now stands at about 90 percent. Patients with matched sibling donors have the best overall prognosis, but a recent analysis reveals 1) comparably high survival rates for boys under age 5 years who receive either an unrelated donor transplant or an HLA identical sibling transplant and 2) a 73 percent five-year survival rate for boys transplanted after age 5 years, suggesting that the transplantation option may be worth considering for those with good clinical status.10
• X-linked lymphoproliferative disease (Duncan syndrome) is a rare PI disorder affecting one to two boys per million. Mortality is high in part due to a unique susceptibility to Epstein-Barr virus, which can result in fulminant fatal disease. Once again, HSCT is the only curative treatment.
A multicenter study of 91 patients — 43 treated with HSCT and 48 not transplanted — documented an overall survival of 81 percent in the HSCT group and 62 percent in the non-transplanted group. Still under debate is whether a newly diagnosed child who is asymptomatic should receive HSCT therapy, as intravenous IG (IVIG) treatment can be instituted promptly; there is general consensus among experts that those with a matched sibling donor should be transplanted.8
Several other PI syndromes for which HSCT has been shown to dramatically improve the prospects for long-term survival include hemophagocytic lymphohistiocytosis (HLH)11 and IPEX (immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome).12,13
Gene Therapy: The Promise of Cures Becomes Reality
Donor HSCT is clearly curative for the majority of infants with SCID. But major problems persist. GVHD and other transplant-related complications commonly occur in the large share of infants for whom there is no fully matched sibling or family donor. Many children continue to have poor B-cell function and remain dependent on long-term IG therapy. Incomplete immune reconstitution following transplantation leaves many children at ongoing risk for serious opportunistic infections; residual immunodeficiency after partially HLA-incompatible HSCT is still responsible for an estimated 30 percent mortality rate at one year post-transplantation.14
For many years, immunologists have appreciated that — if achievable without introducing new health risks — the ideal curative therapy is to harvest bone marrow from the patient and “transduce” his or her own (autologous) HSCs by using special viral “vectors” to insert normal copies of the mutated gene. These normal genes can in turn produce the critical missing functional protein causing the immunodeficiency disorder.
Two small early trials, one in France15 and another in the United Kingdom,16 proved that autologous CD34-positive hematopoietic bone marrow stem cells transduced with a gammaretroviral vector delivering IL2RG and reinfused into patients with SCID-X1 resulted in a sustained restoration of both cellular and humoral immunity.
Unfortunately, less than three years after their triumphant findings were published in Science in 2000, the French investigative team reported two cases of leukemia. Eventually, five of 20 SCID-X1 patients developed leukemia, attributed to “insertional mutagenesis” that resulted when gammaretroviral vectors activated a known T lymphocyte oncogene. In January 2003, the U.S. halted more than two dozen gene therapy studies that utilized those vectors.
Several years later, researchers returned to the clinic with redesigned vectors to ferry functional genes, including novel “self-inactivating” gammaretroviral vectors and lentiviral vectors incorporating safety features intended to minimize the risk of inducing leukemia. A wave of early trial results strongly suggest that gene therapy is safe and curative in patients with SCID-X1 and ADA-SCID.
Gene therapy for SCID-X1. In a report on parallel trials in the U.S. and Europe,17 autologous bone marrow-derived HSCs transduced by a self-inactivating retroviral vector carrying a normal copy of the IL2RG gene were reinfused into nine infant boys with confirmed SCID-X1, including profound deficiency of autologous T cells. All were over age 3.5 months at the time of treatment; the median age was 8 months. All nine patients either lacked an HLA-identical related or unrelated donor or had an active treatment-resistant SCID-related infection. One patient with preexisting severe systemic adenoviral disease died before he could be fully reconstituted with vector-modified CD34-positive HSCs. Another who received a graft with a low number of vector copies did not show evidence of vector DNA uptake and later received a mismatched umbilical cord blood transplant.
At a median follow-up of 29 months, all eight treated boys were still alive, and seven of the eight experienced T-cell proliferative capacity in the normal range, and associated functionality that led to resolution of their infections. No severe adverse events related to the gene-transfer vector or cell manipulations were reported in any of the children. At a median of 33 months of follow-up, there was no occurrence of leukemia. These children are scheduled to be followed and periodically tested over the next 15 years.
Gene therapy for ADA-SCID. In 2009, a multinational team reported that nine of 10 children receiving autologous HSCs transduced with a retrovirus carrying the functional ADA gene experienced immune reconstitution with increases in T-cell count and normalization of T-cell function.18 At a median follow-up of four years, all 10 patients were alive, with no reports of leukemia or other serious adverse outcomes. Eight no longer required ADA replacement therapy. The number of severe infections decreased from 0.93 per 10 person-months before gene therapy to 0.13 following gene therapy; the median number of hospitalization days dropped from 45 to two.
A separate clinical study in the UK subsequently affirmed that, when it works, gene therapy for ADA-SCID resolves the profound T- and B-cell immunodeficiency and appears to all but eliminate the risk of severe opportunistic infections.19 In an extraordinary announcement last November, Dr. David Kohn, the lead investigator of two trials of a gene therapy regimen developed at UCLA, reported that all 18 treated infants with ADA-SCID have been cured.20 The UCLA team plans to seek FDA approval for its gene therapy regimen.
What Comes Next
In this new era of TREC screening, clinicians can now identify infants with SCID at birth and employ HSCT to reconstitute a functional immune system during the critical first few months of life, with improved prospects for partial or complete cures. HSCT is already the gold standard treatment for SCID, but going forward we can expect to see fewer treatment failures and higher overall long-term survival statistics. This experience may additionally help clinicians make further refinements to HSCT therapy for other severe PI disorders, potentially leading to its use in more patients with better outcomes. Remarkably, at the same time, new clinical research suggests that gene therapy is finally poised to realize its curative potential for infants diagnosed with SCID-X1, ADA-SCID and someday, hopefully, other genetically well-characterized PI conditions as well. In the very near future, clinicians may find themselves in the position of making patient-by-patient decisions about when to perform HSCT and when to turn to gene therapy for SCID or certain other severe PI disorders. Given the stakes involved for these precious new beings and their parents, it is a future they can look forward to.