Coagulation, or clotting, factors are the necessary proteins that allow the body to form a blood clot. There are 13 different types of clotting factors in the blood, each designated by Roman numerals I through XIII, that work both dependently and independently of each other in what is known as the clotting cascade (Figure 1).
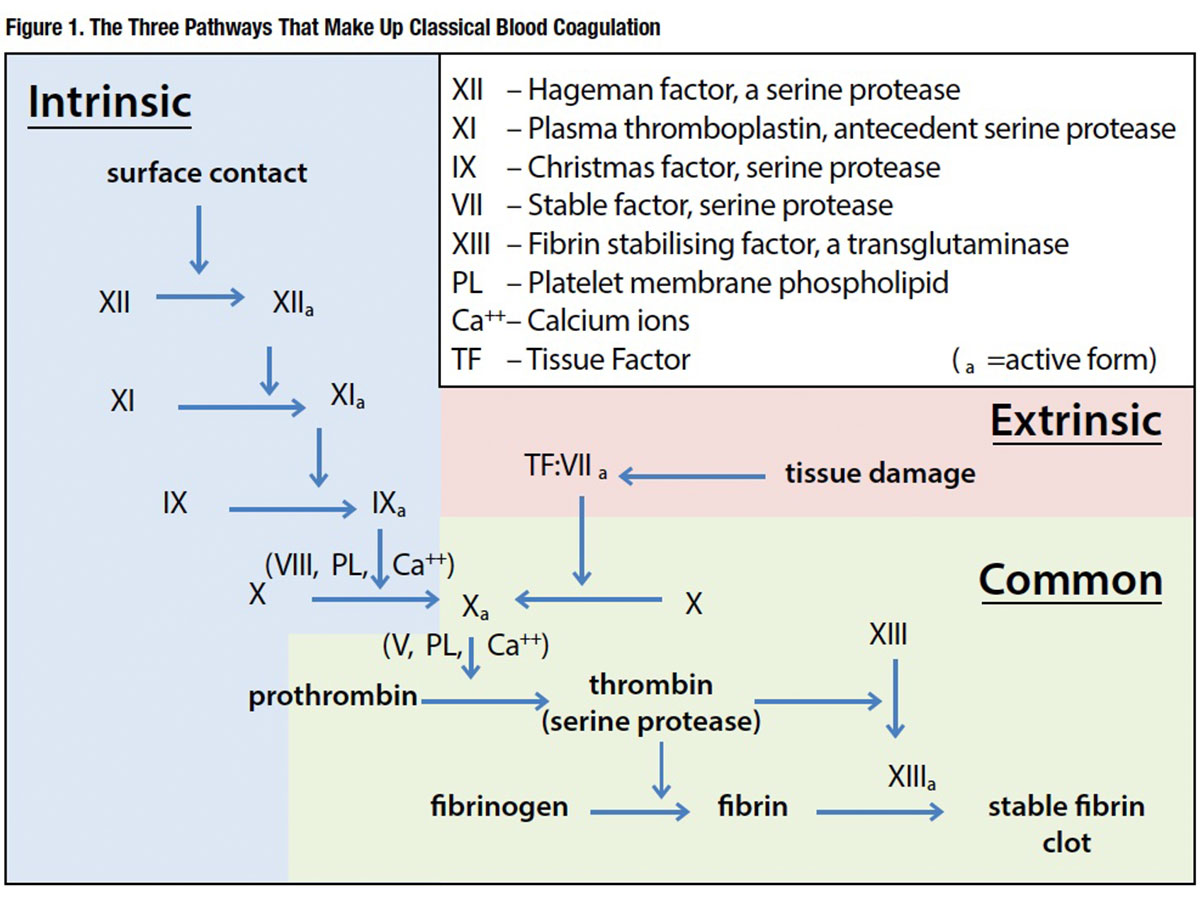
When there is an injury or damage to an area of the body causing bleeding, several things begin to happen almost immediately. The affected blood vessels begin to contract in an effort to slow the blood flow to the area. Platelets begin to stick to the injured space and become “activated” to attract other platelets to the area. Through a process referred to as aggregation and adhesion, the platelets begin to form a plug in the blood vessel hole. It is on the surface of these activated platelets where the clotting cascade occurs and works to form a fibrin clot that will cause hemostasis.
In the event that any one of these factors or components is missing, present in lower than normal levels, or does not work properly, an individual may take longer to stop bleeding and would be diagnosed with a coagulation disorder. There are two types of coagulation factor disorders — hemophilia and von Willebrand disease — that are treated with several different factor replacement products.
Hemophilia
Hemophilia occurs when a person has missing or low levels of FVIII, FIX or, in extremely rare cases, FVII or FXI. It is usually an inherited disease that affects mostly males, although it may occur in people with no family history and also in women. According to the Centers for Disease Control and Prevention (CDC), hemophilia affects one in 5,000 male births, and approximately 400 babies are born with hemophilia each year. Those with hemophilia may experience bruises or bleeds from injuries, or bleeds may be spontaneous events. These bleeding episodes may occur internally in joints, muscles, soft tissues or organs. Untreated or repeated bleeds can cause permanent damage to these areas over time. If the bleeding is uncontrolled or occurs in a vital organ such as the brain, death may occur.
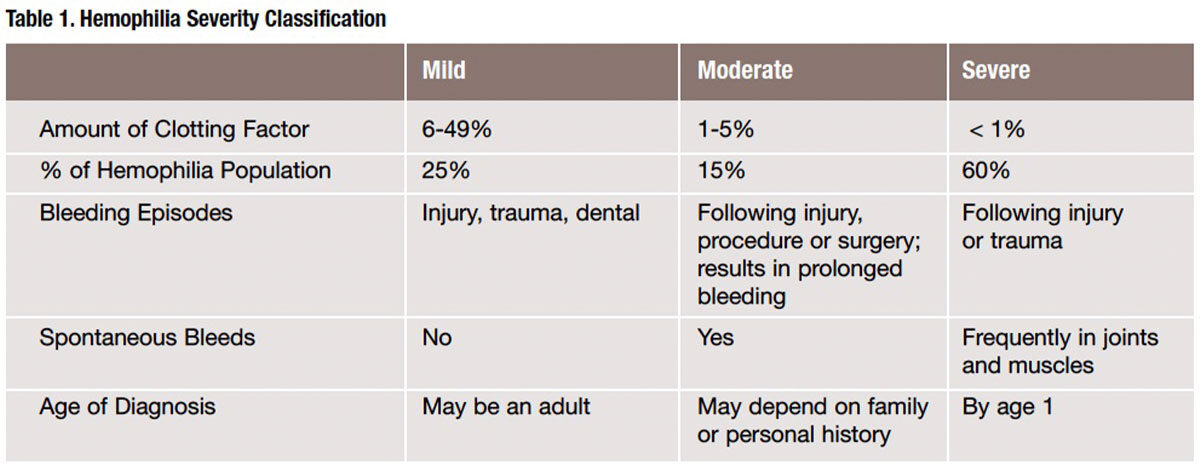
The type of hemophilia a person is diagnosed with is based on the specific factor deficiency that is present and may be further classified by the amount of that clotting factor that is present in the blood. In an individual with normal levels of FVIII and FIX clotting factors, this is 50 percent to 150 percent (Table 1).
While there is no cure for hemophilia at this time, it may be managed with replacing the factor deficiency either prophylactically to prevent bleeding or on demand to treat the bleed. All factor replacement products are given intravenously and, in many cases, by the patients themselves (see Table 2 for a list of factor coagulation products available in the U.S.). Prophylactic therapy is the routine administration of factor one to three times a week. The dose and frequency is based on several patient-specific factors such as age, weight, severity and activity level. The goal of prophylactic factoring is to prevent bleeding episodes from occurring, therefore avoiding many of the associated complications. On-demand or as-needed dosing may be used by all patients to treat an active bleed, or it may be used prior to an event (e.g., medical procedure) that will most likely result in a bleed. An active bleed may require infusions for multiple days or even more than one dose per day to treat.
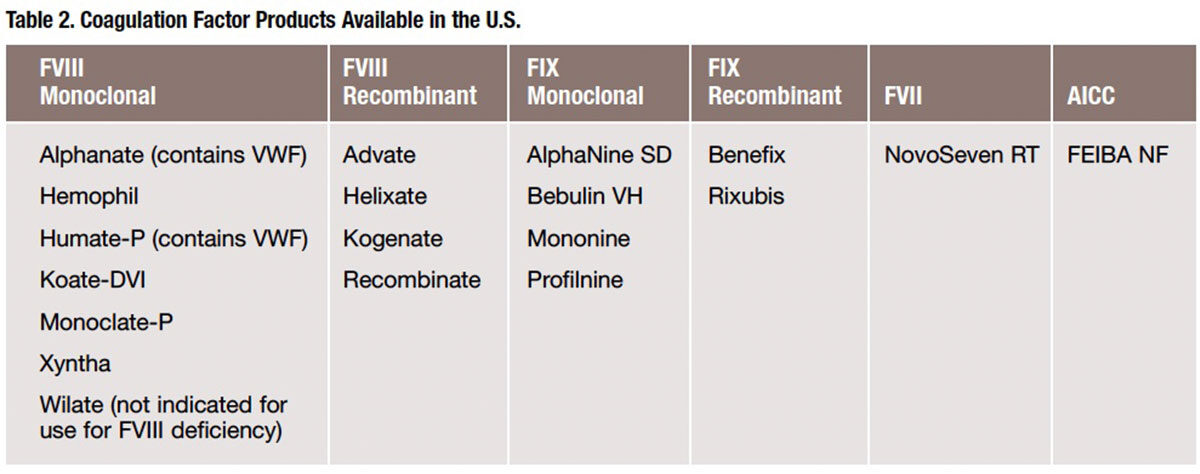
FVIII deficiency, also known as hemophilia A or classic hemophilia, is the most common type of hemophilia as it accounts for 80 percent of all diagnosed patients. FVIII products may be plasma-derived or recombinant. While there are many different FVIII replacement products available, each offering unique characteristics in terms of manufacturing process, diluent volume, accompanying supplies and storage requirements, all may be used for patients with FVIII deficiency and need to be reconstituted prior to administration.
FIX deficiency, also known as hemophilia B or Christmas disease, is much less common than FVIII deficiency, comprising 15 percent of hemophilia patients. FIX products are also available as plasma-derived or recombinant.
It is important to note that patients with FVIII or FIX deficiency must be treated with the specific factor they are lacking for the treatment to be successful. This is because FVIII and FIX are independent clotting factors in the clotting cascade, which means that the action of one is not dependent on the action of the other.
While individuals with FVII deficiency are rare, a FVII replacement product may be used to treat it, as well as FVIII or FIX bleeding disorders. The reason for the latter is because FVIII and FIX are dependent on FVII, which means if the FVII is replaced in the body, it will activate either FVIII or FIX. A FVII replacement product is also useful in the treatment of hemophilia patients who develop inhibitors or antibody to the replacement factor product. Patients with inhibitors are difficult and expensive to manage. Inhibitors destroy the replacement factor product prior to it working, which necessitates much larger or frequent doses of factor products or puts the patient at risk for bleeding episodes. Another alternative treatment in patients with inhibitors is the use of an anti-inhibitor coagulant complex (AICC). This product contains multiple factors (II, VII, VIII, IX and X) that target several areas in the clotting cascade. These multiple sites of action allow for clot formation without the activation of FVIII or FIX, therefore bypassing the inhibitor.
Little is known about bleeding disorders with the remaining clotting factors. This is due in part to their rarity; there may be one in a million in many cases, and they are often caused by an autosomal recessive trait. Also, it’s possible that one of these bleeding disorders may go undiagnosed if patients do not experience a major bleeding episode requiring medical attention during their lifetime. In the event emergency treatment is required, broad-reaching treatments such as fresh frozen plasma, cryoprecipitate or prothrombin complex concentrate (PCC) factor replacement products are typically used. PCCs contain multiple clotting factors along with several proteins, and are used to reverse the anticoagulation effects of warfarin. Currently in the U.S., a three-factor product (II, IX and X) and a four-factor product (II, VII, IX and X) are available; however, they are not used routinely in the treatment of factor disorders due to the high cost, as well as increased risk of thromboembolic events such as pulmonary embolism, deep vein thrombosis, stroke and heart attack. And, even if patients have a positive response to treatment, it becomes difficult to determine the specific factor deficiency that exists, if any, without extensive testing, which may not be widely available.
Von Willebrand Disease
The most common of all coagulation factor disorders is von Willebrand disease (VWD). CDC estimates that this disorder may affect up to 1 percent of the general population. VWD occurs when individuals have lower-than-normal or non-functioning von Willebrand factor (VWF), which is the protein that helps the platelets become sticky early in the clotting process. Unlike hemophilia, VWD is seen in both men and women, although women are more likely to be diagnosed due to increased bleeding during menstrual cycles or childbirth. Because VWF is carried on clotting FVIII, people with VWD may also have lower levels of FVIII.
Like hemophilia, VWD is further classified into three types — 1, 2 and 3 — based on the amount and/or activity level of VWF that is present. It is imperative to know the specific type of VWD a patient has because the treatment varies for each.
Type 1 VWD is the most common and mildest form of the disease and occurs when individuals have normal functioning VWF but lower levels of it. This is most often treated with desmopressin acetate nasal spray as it causes stored VWF to be released into the blood stream. Type 2 VWD occurs when VWF is present in normal levels but does not function properly. The characteristics of dysfunction will further classify type 2 VWD into four categories: 2A, 2B, 2N and 2M. Type 2 VWD may be treated with desmopressin acetate nasal spray or FVIII product containing VWF. Type 3 VWD occurs when there is little or no VWF present. A patient with this type must be treated with a FVIII product containing VWF and may be treated prophylactically. Patients with VWD may also be prescribed aminocaproic acid, which helps maintain clots once formed.
Ongoing Studies in Coagulation Factor Deficiencies
There are many ongoing studies looking into coagulation factor deficiencies, including their causes and symptoms, as well as the products that treat them. A few examples include:
U.S. Food and Drug Administration (FDA) FVIII Study. FDA’s Division of Hematology is responsible for the review of FVIII products used for the treatment of hemophilia A patients. To better characterize these products, it is studying how the structure of FVIII affects the speed with which the liver removes it from the circulation by means of hepatic receptors — proteins on liver cells to which FVIII binds. The results of this study could further improve understanding of the mechanisms of FVIII removal from blood, and facilitate FDA review and approval process for FVIII products, especially of those that are designed to have a long lifetime in the circulation.1
St. Jude Children’s Medical Center FIX Gene Therapy Study. Investigators at St. Jude are testing a novel gene therapy approach to treating and, ultimately, curing severe hemophilia B. In a Phase I/II trial that differs from previous clinical trials of AAV-mediated gene transfer, the researchers “developed a FIX-expression cassette that is packaged as complementary dimers within a single virus particle.… To avoid antibody-mediated immune responses to AAV, the vectors were pseudotyped with capsid of serotype 8 (AAV8), which is less common in humans than AAV2 is and, thus, less likely to stimulate an immune reaction.… [In addition,] AAV8 preferentially targets hepatocytes (liver cells), so the vector can be injected into a peripheral vein, a simple, noninvasive approach that is safe for patients who are prone to bleeding. Once injected, the vector circulates in the blood until it reaches the liver, where it infects hepatocytes and transfers its genetic information.”
Of the first six adult male participants with severe hepatitis B to participate in the trial, five received regular FIX prophylaxis two to three times per week before gene transfer. Participants were enrolled sequentially into one of three dose cohorts (low, intermediate and high). After gene transfer, all six participants showed an increase in FIX levels that was roughly vector dose-dependent. Three of the six participants no longer require FIX concentrate prophylaxis to prevent spontaneous hemorrhage, and three have been able to extend the interval between prophylaxes. These participants had severe hemophilic arthropathy before they entered the study. Because of the results of this study, future efforts will include accruing four more participants, following each participant for a longer period to fully define the benefits and risks of this therapy, and optimizing vector dosing.2
CDC Inhibitor Project. CDC is looking into how and why about one-third of those who take blood products to correct coagulation FVIII and FIX deficiencies develop inhibitors, creating far greater difficulty in treating their condition, as well as greatly increasing therapy costs. The goal of the study is to determine whether a change in treatment products (from one type of factor product to another) leads to an inhibitor, whether people with specific gene mutations are more likely to develop an inhibitor, what characteristics make some people more likely to develop an inhibitor than others, why some people develop inhibitors and others do not, and how often inhibitors occur. It is hoped that learning more about why some people develop inhibitors and others do not may help to predict who will develop an inhibitor before treatment is started, which may lead to a decreased rate of inhibitors, decreased healthcare costs, and the licensure of safe and more effective treatment products for people with hemophilia.3
Hemophilia: Today and Beyond
It is estimated that the number of new cases of coagulation factor disorders in the world will likely remain constant. However, people with these disorders are living longer today than ever before, and current treatments are both effective and safe. Nevertheless, researchers are looking for better forms of treatment. A great deal of progress has been made over the past 50 years, and now that advances are being made in the fields of biotechnology and gene therapy, it’s certainly plausible that there could be a long-term cure for the disease.