In This Article:
The major challenges of current [hemophilia] treatment regimens, such as the short half-life of therapeutics with the
need for frequent intravenous injections, encourage the current efforts to produce coagulation factors with more
prolonged bioavailability.
— Massimo Franchini and Pier Mannucci (2012)
Over the last 50 years, perhaps no serious chronic health disorder has undergone a more radical transformation in its management and long-term prognosis than hemophilia. Prior to the discovery of cryoprecipitate in 1965 and availability of the first factor concentrates a few years thereafter, persons with severe hemophilia spent much of their lives in hospital wards and rehabilitation services; many died in childhood or early adulthood from uncontrollable hemorrhage in the brain or other vital organs. Those who survived endured extremely painful bleeds into the muscles and joints, particularly the ankle, knee and elbow. For children with severe hemophilia A or B, participation in active sports was out of the question. In surviving adults, the sequelae of years of recurrent hemarthroses could be seen on an x-ray or from across a room: joint deformity, degenerative arthritis, flexion contractures and restricted mobility.
Replacement therapy made possible by the introduction of factor VIII and IX concentrates in the late 1960s drastically reduced mortality risk, limited the severity of damage caused by hemorrhages into joints, tissues and vital organs, and freed patients to travel, hold steady jobs and lead near-normal lives. But over the 25 years that followed, the U.S. standard treatment paradigm — reactive “on-demand” self-administration of clotting factor upon awareness of a developing bleed — continued to translate into emergency room visits, hematomas and hemarthroses, major risk of joint damage, and restricted active play and sports participation.
Prophylaxis: The Next Leap Forward
Since 1958, Swedish boys with severe hemophilia have received continuous prophylaxis, beginning in infancy, in an attempt to proactively convert the disease to a milder form and minimize hemophilic arthropathy. In numerous published reports, Swedes and other European treaters showed that primary prophylaxis prevents both crippling joint damage and disabling or fatal brain hemorrhage. Initiating prophylaxis in very young children, before they experience their first joint bleeds, has been shown to be the most effective strategy to reduce later arthropathy risk.
Finally in 1994, the U.S. National Hemophilia Foundation (NHF) issued a new recommendation encouraging physicians to consider routine prophylactic infusions of appropriate clotting factor to prevent bleeding episodes. In 2007, NHF expanded its recommendation to advise that “prophylaxis using the following regimen be considered optimal treatment for any individuals with severe hemophilia A or B: 25-50 factor VIII units/kg three times per week or every other day, and 40-100 factor IX units/kg two to three times weekly.”
The emergence of routine prophylactic replacement as the standard of care for children and adolescents with severe hemophilia has also introduced a major new challenge: treatment compliance. Because of the very short intravascular half-life of administered factor concentrates — about 12 hours for factor VIII and 18 hours for factor IX — the product must be administered twice weekly to as often as every other day. It is inconvenient, time-consuming and unpleasant. Compliance can be an issue in particular for teenage boys responsible for performing their own injections, who may become complacent or simply forget in the course of their busy lives. In some children, frequent regular dosing may also necessitate placement and use of central venous access devices, accompanied by risk of significant medical complications that can include infections, sepsis and thrombosis.
The obvious solution was to develop and introduce coagulation factors that persist longer in the circulation, requiring less frequent injections.
Extended Half-Life: The Newest Leap Forward
After years of anticipation, the first of what promises to be a number of bioengineered products featuring extended half-life have finally been approved for marketing, with several others awaiting regulatory approval. These products offer not only the advantage of less-frequent injections and the prospect of improved treatment compliance, but recent evidence suggests the additional benefit of a reduced number of follow-up injections needed to support complete healing following episodic bleeds.
Recognition that the pharmacokinetics of these novel clotting factors can vary widely from one person to the next has added impetus to another important advance in hemophilia treatment: individualized therapy. With individualized therapy, infusion frequency and dosage are guided by 1) trial dosing with the extended half-life product to ascertain the patient’s pharmacokinetic profile, 2) the severity of his factor deficiency, 3) his bleeding pattern, 4) the condition of his musculoskeletal system and 5) his level of physical activity.
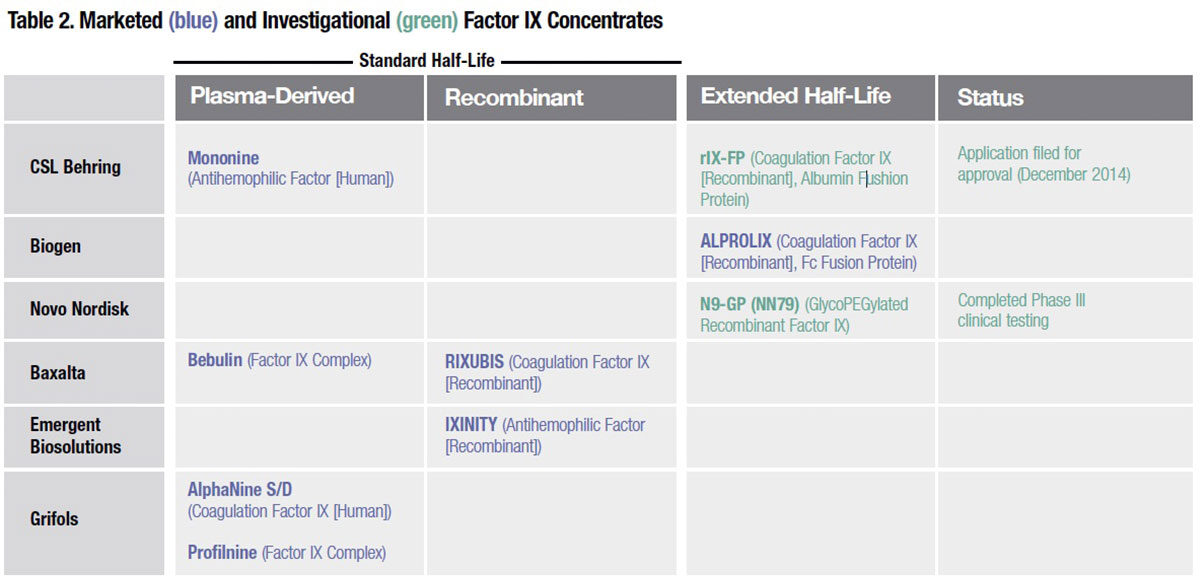
First to market: Fc fusion proteins. Biogen was first to reach the market last year with ELOCTATE (antihemophilic factor [recombinant], Fc fusion protein) and ALPROLIX (coagulation factor IX [recombinant], Fc fusion protein). The Fc fragment bound to each of these clotting factors exploits the same natural mechanism that protects immunoglobulins from rapid lysosomal degradation following endocytosis by vascular endothelial cells. In essence, recognition of the Fc fragment causes these proteins to be “cycled” instead back into the circulation. The result is circulating half-life that, on average, is increased in relation to conventional factor VIII and IX by 1.8-fold for ELOCTATE and by at least four-fold for ALPROLIX.
In the pivotal clinical trial of ELOCTATE, the treatment interval and dose were individualized to maintain trough levels between 1 percent and 3 percent above baseline or higher, as clinically indicated to prevent bleeding. Among 112 subjects, 111 achieved a dosing interval of three days or longer; ultimately, nearly 30 percent were managed with a dosing interval of five days or longer. With a mean half-life exceeding 80 hours, ALPROLIX can be administered every seven to 10 days — dramatically reduced from the usual standard of two to three times weekly.
PEGylated and glycoPEGylated products. Already proven as a means to extend the half-life of more than a dozen licensed proteins and peptides, PEGylation involves the attachment of long strands of polyethylene glycol (PEG) to selected locations on the therapeutic protein of interest. PEGylation has been applied to a total of four investigational recombinant factor VIII and IX products with the goal of increasing their circulating half-life.
Of the four extended half-life factor VIII products currently in late-stage development, three utilize PEGylation or glycoPEGylation, a variation wherein glycans present on the protein are modified to allow site-specific conjugation of PEG (Table 1). It is thought that the long, constantly moving strands of PEG act to protect the factor VIII protein against immune cells, antibodies, enzymes and other blood constituents that normally attach to it and remove it from the circulation. A glycoPEGylated recombinant factor IX developed by Novo Nordisk (N9-GP) is also in late-stage clinical testing (Table 2). But working with PEGylation technology is not without its own risks, as Novo Nordisk learned when a case of hypersensitivity and a lack of dose-response linearity prompted the company to discontinue development of an investigational glycoPEGylated factor VIIa intended for use in inhibitor patients.
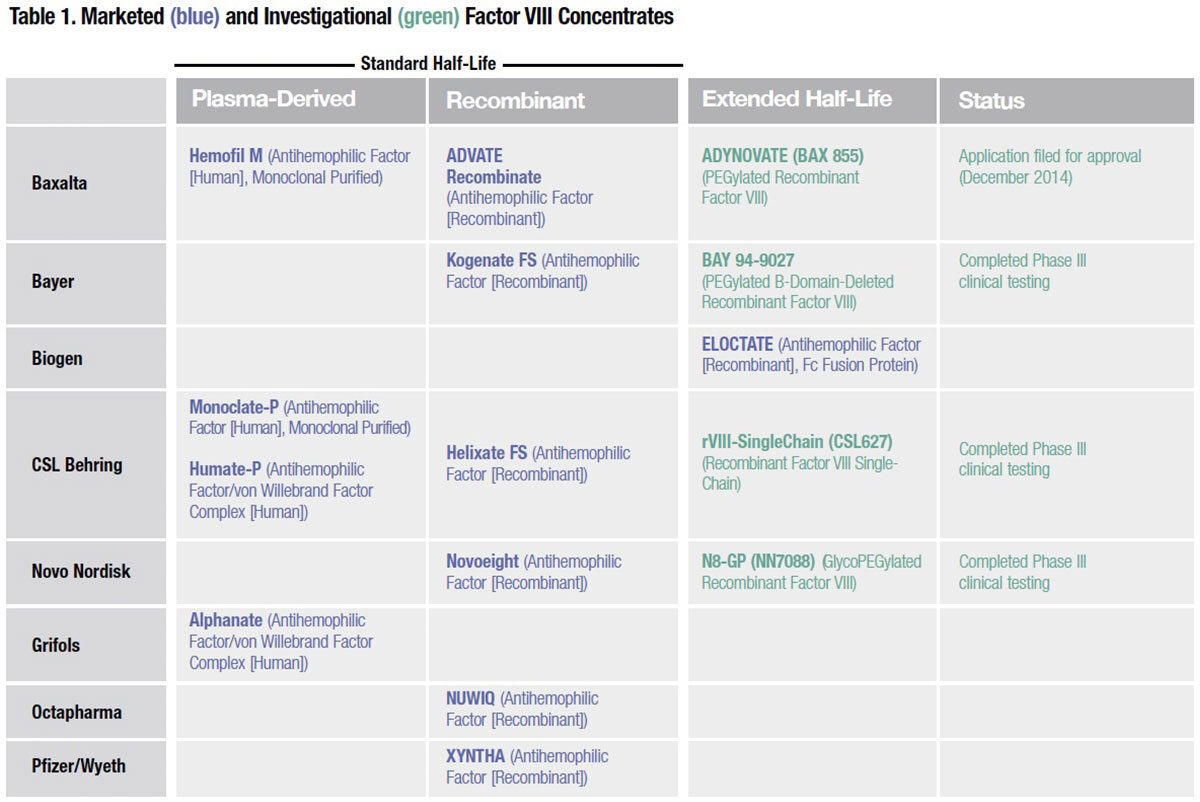
Single chain factor VIII. CSL Behring is investigating a recombinant factor VIII molecule that exploits the natural stabilizing effect of von Willebrand factor (VWF) to extend half-life. Its single-chain recombinant factor VIII (rFVIIISingleChain) has a strong affinity for VWF, resulting in enhanced stability and integrity of the protein in circulation. In a prophylaxis study evaluating rFVIII-SingleChain, 32 percent of subjects could be dosed weekly, while another 54 percent were dosed three times per week — again illustrating how individual variability in pharmacokinetics, as well as other risk parameters, can influence bleeding tendency from one patient to the next.
As with other extended half-life factor VIII products that have been evaluated in Phase III studies, there is no evidence of inhibitor antibody development following thousands of infusions of rFVIII-SingleChain.
Albumin fusion proteins. Capitalizing on the long intravascular half-life of human albumin — about 20 days — CSL Behring has designed a novel investigational fusion protein that links it to recombinant factor IX (rIX-FP). Like the Fc portion of IgG1, the albumin bound to factor IX is unlikely to elicit a neutralizing antibody response. Another appeal is that albumin fusion products can be manufactured with fewer post-expression modifications and purification steps than PEGylation, and more efficiently in relation to other fusion protein approaches, including use of the IgG1 Fc fragment.
CSL Behring is evaluating multiple prophylaxis regimens, including seven-day and even 14-day intervals, in a Phase II/III safety, pharmacokinetic and efficacy study of previously treated patients with hemophilia B and baseline factor IX of ≤2 percent. In February, the U.S. Food and Drug Administration (FDA) accepted the company’s application for approval of rIX-FP, which is currently under review.
While average 1.5- to 1.7-fold increases in circulating halflife have been reported for ELOCTATE and all of these novel long-acting factor VIII candidates, to about 18 to 20 hours, long-acting factor IX products perform far better. Together with ALPROLIX, four- to five-fold increases in mean half-life have been documented in pharmacokinetic studies of both Novo Nordisk’s glycoPEGylated factor IX product (N9-GP) and CSL Behring’s factor IX-albumin fusion protein (rIX-FP).
Also in development by CSL Behring is an investigational anti-inhibitor product based on this same platform: a recombinant fusion protein linking factor VIIa with albumin (rVIIa-FP) for on-demand treatment of patients with congenital hemophilia A or B who have developed inhibitor antibodies to factor VIII or IX replacement therapy. In August of this year, the first patient was enrolled in a Phase II/III study evaluating its pharmacokinetics, efficacy and safety.
Innovations Targeting Other Bleeding Disorders
The current wave of product innovation has produced new treatment options for other important coagulation disorders beyond hemophilia A and B. Below are two products newly licensed and available within just the last two years:
- OBIZUR (antihemophilic factor [recombinant], porcine sequence) (Baxalta) for treatment of acquired hemophilia A; approved October 2014. Baxalta plans to submit additional clinical trial data to support an indication for perioperative management of bleeds in adults with acquired hemophilia A.
- TRETTEN (coagulation factor XIII A-subunit [recombinant]) (Novo Nordisk) for treatment of congenital factor XIII A-subunit deficiency; approved December 2013.*
In addition, an application for marketing approval of the first recombinant von Willebrand factor, Baxalta’s VONVENDI (BAX 111) was submitted in late 2014, and is currently under review by FDA. Phase III clinical testing is now in progress to evaluate its efficacy and safety in patients with von Willebrand disease undergoing surgery.
Design Better Products, Patients Come
When allowed to choose between continuing on an extension study with investigational extended half-life products or returning to treatment with their previous standard half-life product, all subjects who have participated in completed Phase III trials have chosen to continue with the long-acting product. This strong preference for a product that reduces the number of factor infusion sessions and needle sticks may translate into improved treatment compliance. Better compliance and more days of protection with each administration should mean fewer days during the year that factor trough levels fall below the target level. And that, in turn, should mean fewer breakthrough bleeding events.
For persons with hemophilia, all evidence suggests that a healthier, safer future has indeed arrived.
* CSL Behring’s Corifact factor XIII concentrate (human), indicated for routine prophylactic treatment and perioperative management of surgical bleeding in adult
and pediatric patients with congenital factor XIII deficiency, was approved in February 2011.